電荷移動錯体
電荷移動錯体(でんかいどうさくたい、英: Charge-transfer complex、略称: CT錯体)あるいは電子受容-供与錯体(英語: Electron-donor-acceptor complex、略称: EDA錯体)とは、電荷が分子間で移動できる2つ以上の異なる分子もしくは1つの巨大分子の異なる部分の会合体である。会合により分子が静電気的に引きつけられ、錯体が安定化される力が生まれる。電子を供与する分子は電子供与体、電子を受容する分子は電子受容体と呼ばれる。
電荷移動錯体における静電気的な結合は安定なものではないため、共有結合よりずっと弱い。多くの錯体は励起状態で電荷移動遷移を引き起こす。これらの錯体は電子のエネルギーが変化する際に電磁スペクトルにおける可視光領域の光と同じエネルギーを吸収するため、特有の色を持つ。この吸収帯は電荷移動吸収帯(CT帯)と呼称される。スペクトルを測ることで電荷移動吸収帯を決定できる。
電荷移動錯体は無機分子や有機分子、固体や液体に溶液と様々な種類が存在する。よく知られているのはヨウ素がデンプンと会合して青紫色になる反応である。
無機化学では、ほとんどの電荷移動錯体が金属原子と配位子の間での電子移動によって成り立っている。遷移金属錯体の電荷移動吸収帯は金属と配位子、それぞれの分子軌道間での電荷密度の移動に起因している。配位子から金属への電荷移動が起こる錯体をLMCT錯体、金属から配位子への電荷移動が起こる錯体をMLCT錯体と呼ぶ。したがって、MLCT錯体は中心金属を酸化し、LMCT錯体は還元する。共鳴ラマン分光もこれらの電荷移動吸収帯の特定に用いられる[1]。
目次
1 供与と受容の平衡
2 電荷移動遷移エネルギー
3 電荷移動吸収帯の特定
4 無機電荷移動錯体
4.1 LMCT
4.1.1 LMCTのエネルギー傾向
4.2 MLCT
4.2.1 MLCTの励起状態における光反応性
4.3 電荷移動錯体の色
5 その他の例
6 電導性
7 脚注
8 関連項目
供与と受容の平衡
電荷移動錯体は分子、もしくは分子の一部が弱い結合でつながってできており、一方が電子供与体、他方が電子受容体として働く。結合は共有結合のように強くはないため、温度や濃度、溶媒などの環境に左右される。
電荷移動錯体では電子供与体(D)と電子受容体(A)の分子が次のような平衡を成り立たせている:
- D+A↽−−⇀DA{displaystyle {ce {D + A <=> DA}}}

量子化学では、非結合状態 |D, A> と配位状態 |D+...A−>の共鳴で表現される。配位状態になるために、電子遷移によって吸収帯が可視光領域になる。
吸収スペクトルにおける電荷移動吸収帯の強さは会合反応の平衡定数に大きく依存する。錯体の会合定数(解離定数の逆数)を決定する方法として、受容体と供与体の濃度が既知の溶液の吸収の強さを調べる方法が知られている。この方法はヨウ素と、溶媒の芳香族炭化水素の会合定数を調べる方法として最初に発表されている[2]。この方法は、発表した人の名前にちなんでベネシ・ヒルデブランド法と呼ばれる。
電荷移動遷移エネルギー
電荷移動遷移エネルギーは各錯体に特有であるため、各錯体が吸収する波長もまた各錯体に特有である。
供与体分子の電子供与力はイオン化ポテンシャル(最高被占軌道から電子を1個取り除くのに必要なエネルギー)によって測ることができる。また受容体分子の電子受容力は電子親和力(最低空軌道に1個電子を入れる際に放出されるエネルギー)によって決定される。
全体のエネルギー収支 (ΔE) の値は自発的な電荷移動によって発生するエネルギーの値から得られる。エネルギー収支は受容体の電子親和力 (EA) と供与体のイオン化エネルギー (EI) の差に受容体と供与体の間の静電気的エネルギーを足して得られ、以下の式で表現できる[3]。
- ΔE=EA−EI+J{displaystyle {Delta }E=E_{A}-E_{I}+J,}

電磁スペクトルにおける電荷移動吸収帯の位置はこのエネルギー差と共鳴における非結合状態と配位状態の寄与のバランスに密接に関わっている。
電荷移動吸収帯の特定
電荷移動錯体は以下のように特徴付けられる[1]。
- 色
- 電荷移動錯体の色は供与体から受容体への電荷移動によって生じるエネルギー差を反映している
- ソルバトクロミズム
- 溶液では遷移エネルギーや錯体の色が溶液の比誘電率によって変化し、電荷遷移にバリエーションが生まれる。これは配位子のπ→π*遷移とは区別される。
- 吸収の強さ
- 電荷遷移吸収帯は紫外可視領域にあることが多い。無機錯体では、モル吸光係数εが約50000 L mol−1 cm−1にもなり、一般的なdd遷移(t2g軌道からeg軌道への遷移)のモル吸光係数(ε=20 L mol−1 cm−1以下)の千倍以上にもなる。これは電荷移動遷移がスピン許容かつラポルテ許容だからである。一方d-d遷移はスピン許容である場合があるが、常にラポルテ禁制である。
無機電荷移動錯体
電荷移動は金属が関わる錯体の無機配位子内で起こる。電荷移動の方向によってLMCTとMLCTに分類される。
LMCT
LMCT錯体は配位子性の強い分子軌道から金属性の強い分子軌道への電荷の移動によって生じる錯体である。このタイプの遷移は錯体が比較的エネルギーの高い孤立電子対を持っていたり(SやSeなど)、金属に低位の空軌道があったりする場合に起きやすい。このような錯体の多くは金属の酸化数が高い(d0の場合も存在する)。これらの配位化合物は受容体が低エネルギーでも電子を受け入れられることを意味している。
IrBr63−などのようにd6電子を持つ正八面体型錯体はt2g軌道が埋まっている。結果として、吸収の極大は配位子のσ軌道から空のeg軌道に遷移するエネルギーに対応する250 nm付近の紫外線領域に生まれる。しかしd5電子を持つIrBr62−錯体は2つの吸収帯をもつ。一つは600 nmでもう一つは270 nmである。これは一つがt2g軌道(電子をもう一個入れることができる)でもう一つがeg軌道への吸収に対応している。600 nmの吸収帯はt2g軌道に、270 nmの吸収帯はeg軌道に対応する。
電荷移動吸収帯は非結合性軌道から配位子のeg軌道への吸収によって発生する場合もある。
LMCTのエネルギー傾向
- 酸化数
- +7 MnO4− < TcO4− < ReO4−
- +6 CrO42− < MoO42− < WO42−
- +5 VO43− < NbO43− < TaO43−
遷移のエネルギーはイオン化傾向と相関がある。もっとも還元されやすい金属イオンがもっとも遷移エネルギーが低くなる。この傾向はLMCT全体に対して共通であるため、金属イオンは配位子によって還元されることになる。
例:
- MnO4− : 過マンガン酸塩は四面体形分子構造をしているため電子が満たされている酸素原子のp軌道からマンガン(VII)イオンの空の軌道へと電子が遷移する移動が起きやすいため、紫色に強く発色する。
CdS: カドミウムイエローの色はS2−のπ電子がCd2+の5s軌道に移るために生じる。 .
HgS: S2−のπ電子がHg2+の6s軌道に移るために赤い色を生じる(辰砂)。- 鉄の酸化物: O2−のπ電子が鉄の3d軌道に移るために赤や黄色になる。
MLCT
MLCT錯体は電子が金属から配位子に移ることで生じる[1][4] 。このような錯体は配位子が低エネルギーのπ*軌道を持つ芳香族化合物である場合によくみられる。電子の移動は金属の酸化数が比較的低く、軌道のエネルギーが高い際に見られる。
MLCTを形成する配位子には2,2'-ビピリジン(bipy)、1,10-フェナントロリン(phen)、CO、CN−やSCN−などがある。以下にこのような錯体の例を示す。
トリス(ビピリジン)ルテニウム(II)塩化物 : オレンジ色の錯体である。励起状態は電荷移動によって発生し、マイクロ秒の寿命を持つため、光化学酸化還元反応の多目的反応剤としても研究されている[5]。- W(CO)4(phen)
- Fe(CO)3(bipy)
MLCTの励起状態における光反応性
MLCT錯体の光反応性は酸化された金属と還元された配位子の性質によって生まれる。トリス(ビピリジン)ルテニウム(II)塩化物やRe(bipy)(CO)3Clといった一般的なMLCT錯体はあまり反応しないが、光反応性を持つMLCT錯体が数多く合成されている。
フォーグラー (Vogler) とカンケリー (Kunkely) はMLCT錯体が酸化された金属と還元された配位子からなる基底状態の異性体であると考えた[6]。したがって、還元された配位子での求電子攻撃やラジカル反応、金属中心での酸化的付加、外圏電子移動などの様々な反応は、MLCT電子移動によって生じる状態変化に起因すると考えたのである。MLCT状態の反応性は多くの場合金属の酸化状態に依存する。全体的な反応プロセスとしては会合的な配位子の置換反応、 エキサイプレックスの生成、そして金属結合の開裂からなる。
電荷移動錯体の色
図. 1 I2•PPh3の電荷移動錯体をCH2Cl2に溶かしたときの溶液の色。左から順に:
(1) I2 dissolved in dichloromethane - 電荷移動錯体は生成しない
(2) 過剰のPPh3を加えて数秒後。電荷移動錯体が生成している。
(3) 過剰のPPh3を加えて1分後、電荷移動錯体[Ph3PI]+I−が生成している。
(4) 過剰のI2を加えた直後。[Ph3PI]+[I3]−が生成している[7]。
多くの金属錯体はd-d遷移により特有の色を持つ。可視光線から錯体に特有の波長の光を吸収するとd電子が励起される。この光の吸収により色が生まれる。この色は通常かなり弱い。これは選択則による。
- スピン則: Δ S = 0
遷移の際、電子のスピンが変化するのは好ましくない。スピンが変化する反応はスピン禁制反応と呼ばれる。
ラポルテの規則: Δ l = ± 1
対称中心を持つ錯体におけるd-d遷移は禁制である。これは「対称禁制」や「ラポルテ禁制」に相当する[8]。
電荷移動錯体はd-d遷移を起こさない。したがってこれらの規則は適用されず、非常に強い吸収が見られる。
例えば、昔から知られているデンプンから形成される電荷移動錯体は青紫色になる。これは贋金の識別に利用された。アメリカの紙幣は普通の紙と異なりデンプンが含まれていなかった。そのため、もし紙幣をヨウ素溶液に浸して紫色になれば、それはデンプンが含まれている紙であるためニセ金と判断できた。
その他の例
Hの形をしたヘキサフェニルベンゼン (Fig. 2) は電子を自己供与して電荷移動錯体を形成する。この錯体をサイクリックボルタンメトリーにかけるとH+からH4+に相当する4つのピークが検出される。この時の第1イオン化エネルギーE1/2は0.51 eVにすぎない。ゆえにこれらの芳香族炭化水素はドデカメチルカルボニル ('B’で表す)によって酸化されて青い結晶(H+B-)になりやすい[9]
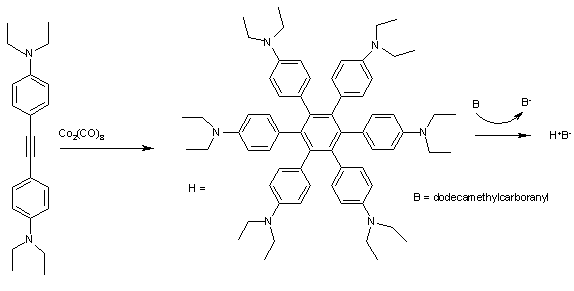
Fig. 2 H+B−錯体の合成: オクタカルボニル二コバルトを触媒とする二置換アルキンのアルキン三量化が起こる。このときジ(エチルアミノ)基などの電子供与基などによって電子が非局在化されるのが望ましい。
フェニル基は全て中心の芳香環と45°の角度をなして配座しており、ラジカルカチオンの正電荷は環状になっている6つのベンゼン環で共有されて、全体で非局在化している。この錯体は近赤外線領域に5つの吸収帯をもち、デコンボリューションとマリケン・ハッシュ理論を使ってそれぞれの吸収が特有の分子電子遷移に割り当てられる。
電導性

ヘキサメチレンTTF/TCNQ 電荷移動錯体の結晶構造の側面図。層の端にある原子を強調している[10]。

ヘキサメチレンTTF/TCNQ 電荷移動錯体の結晶構造の端面図。TTFの層の間の距離は3.55 Åである。
1954年、ペリレンとヨウ素もしくは臭素を組み合わせた電荷移動錯体が合成され、電気抵抗率が8 Ω・cmまで低下することが報告された[11][12]。1962年には、現在はよく知られた電子受容体であるテトラシアノキノジメタン (TCNQ) が報告された。テトラチアフルバレン (TTF)は1970年に報告され、強い電子供与体であることがわかった。1973年にはこれらの化合物の組み合わせによって強い電荷移動錯体が生成することが発見された。この錯体はTTF-TCNQと呼ばれている[13]。この錯体は溶液中で生成し、結晶化も可能であることがわかった。結晶はほぼ金属のような電気伝導性を示し、最初の有機伝導体として報告された。TTF-TCNQの結晶中ではTTFとTCNQの分子が独立かつ平行に配置されており、電子遷移は供与体 (TTF) の並びから受容体 (TCNQ) の並びへと起こる。ゆえに電子とホールが別々に存在し、それがTCNQの層とTTFの層でそれぞれ縦に並ぶため、電子はこの層を突き抜ける筒の中を通るように動く。この電子のポテンシャルは結晶の端にある並びで決まる。
テトラメチル-テトラセレナフルバレン-ヘキサフルオロリン酸塩錯体(TMTSF2PF6)は室温では有機半導体であるが、転移温度0.9 K、圧力12 kbarで有機超伝導体に変化する。残念ながら、この錯体の臨界点での電流密度は非常に小さい。
脚注
- ^ abcピーター・アトキンス; Shriver, D. F. (1999). シュライバー・アトキンス無機化学(下) (第6版 ed.). 東京化学同人. p. 634-636. ISBN 978-4-8079-0899-8.
^ H. Benesi, J. Hildebrand (1949). “A Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons”. J. Am. Chem. Soc. 71 (8): 2703–2707. doi:10.1021/ja01176a030.
^ Mulliken, R. S.; Person, W. B. (1969). Molecular Complexes. ニューヨーク・ロンドン: Wiley-Interscience. Bibcode 1971JMoSt..10..155B. doi:10.1016/0022-2860(71)87071-0. ISBN 0-471-62370-9.
^ Tarr, Donald A.; Miessler, Gary L. (1991). Inorganic Chemistry (2nd ed.). イングルウッド・クリフ (ニュージャージー州): Prentice Hall. ISBN 0-13-465659-8.
^ Kalyanasundaram, K. (1992). Photochemistry of polypyridine and porphyrin complexes. Boston: Academic Press. ISBN 0-12-394992-0.
^ Vogler, A.; Kunkely, H. (2000). “Photochemistry induced by metal-to-ligand charge transfer excitation”. coordination chemistry reviews 208: 321. doi:10.1016/S0010-8545(99)00246-5.
^ Housecroft, C. E.; Sharpe, A. G. (2008). Inorganic Chemistry (3rd ed.). Prentice Hall. p. 541. ISBN 978-0131755536.
^ Robert J. Lancashire. “Selection rules for Electronic Spectroscopy”. 西インド諸島大学. 2008年8月30日閲覧。
^ Duoli Sun; Sergiy V. Rosokha; Jay K. Kochi (2005). “Through-Space (Cofacial) -Delocalization among Multiple Aromatic Centers: Toroidal Conjugation in Hexaphenylbenzene-like Radical Cations”. アンゲヴァンテ・ケミー 44 (32): 5133–5136. doi:10.1002/anie.200501005.
^ D. Chasseau; G. Comberton; J. Gaultier; C. Hauw (1978). “Réexamen de la structure du complexe hexaméthylène-tétrathiafulvalène-tétracyanoquinodiméthane”. Acta Crystallographica Section B 34: 689. doi:10.1107/S0567740878003830.
^ Y. Okamoto and W. Brenner Organic Semiconductors, Rheinhold (1964)
^ H. Akamatsu, H. Inokuchi, and Y.Matsunaga (1954). “Electrical Conductivity of the Perylene–Bromine Complex”. ネイチャー 173 (4395): 168. Bibcode 1954Natur.173..168A. doi:10.1038/173168a0.
^ P. W. Anderson; P. A. Lee; M. Saitoh (1973). “Remarks on giant conductivity in TTF-TCNQ”. Solid State Communications 13: 595–598. Bibcode 1973SSCom..13..595A. doi:10.1016/S0038-1098(73)80020-1.
関連項目
- 有機半導体
- 有機超伝導体


